Results of a retrospective, real-world analysis determined that many patients with chronic myeloproliferative neoplasm (MPN) polycythemia vera (PV) received underdosed hydroxyurea, which led to lower complete response and toxicity rates, as well as proceeded with hydroxyurea regardless of poor clinical or hematological responses. These results highlight the need for more optimized [hydroxyurea] management and intervention in less-than-optimal responders.
“Hydroxyurea is currently the most used cytoreductive therapy for PV patients at high risk of thrombosis, with most patients achieving adequate control of the disease with acceptable tolerance,” Francesca Palandri, MD, Universitaria di Bologna, Bologna, Italy, and coauthors explained. “However, many patients may only obtain a poor response to [hydroxyurea] or develop drug-related toxicities during therapy.”
This study sought to define clinical characteristics associated with the achievement of complete response (CR) to hydroxyurea, investigate whether the type of suboptimal response may influence a decision to switch to ruxolitinib, and assess if a patient achieving CR to hydroxyurea would improve outcome parameters. The analysis included 563 patients with polycythemia vera treated with hydroxyurea for ≥12 months during an observational “PV-NET” Italian study. Among this patient population, 166 patients achieved complete response (CR), 264 achieved partial response (PR), and 133 achieved no response (NR).
In a multivariate analysis, the absence of splenomegaly (P = 0.03), pruritus (P = 0.002), and a median hydroxyurea dose of ≥1 g/day (P < 0.001) remained associated with CR. Overall, 283 patients who received either CR or PR continued hydroxyurea, and 114 patients switched to ruxolitinib. It was noted that many patients continued hydroxyurea despite a PR/NR and that splenomegaly and other symptoms were the main drivers of an early switch.
A median hydroxyurea dose of ≥1 g/day was correlated with more frequent adverse events. In the 449 patients who were receiving only hydroxyurea, rates of thrombosis, hemorrhages, progression, and overall survival (OS) were comparable among the CR, PR, and NR groups.
Palandri et al concluded, “Better [hydroxyurea] management, standardization of the criteria for and timing of responses to [hydroxyurea], and adequate intervention in poor responders should be advised.”
Patients with hematologic myeloproliferative neoplasms (MPNs)—a group of rare blood diseases that include primary myelofibrosis, essential thrombocythemia (ET), and polycythemia vera (PV)—should be more active in their treatment plan, according to experts in oncology pharmacy who participated in a Pharmacy Times clinical forum in Chicago, Illinois, in June 2023. “Our role as pharmacists is to give [patients] as much information as we possibly can, then encourage them to move forward with advocating for themselves,” said Krystal Preston, PharmD, BCPS, a senior clinical oncology pharmacist at CVS Health and a clinical pharmacist at the University of Chicago Medicine.
Patients who are serious about taking an active role in their treatment could inspire health care providers to collaborate more, both with them and with other members of the care team, according to discussion leader Zahra Mahmoudjafari, PharmD, MBA, BCOP, FHOPA, clinical pharmacy manager of hematology, blood and marrow transplant, and cellular therapy at the University of Kansas Health System in Mission.
Subtypes of MPNs, ET, and PV typically transform into myelofibrosis, which can subsequently turn into acute myelocytic leukemia (AML). At least 20% of MPNs may transform into AML; therefore, the goal for treatment is to prevent this from occurring, Mahmoudjafari explained.
With more than 90% of patients with PV having a JAK2 mutation, “it is probably, by far, the mutation that we have the most actionable ability to do something about,” Mahmoudjafari said. She noted that there are 3 FDA-approved Janus kinase (JAK) inhibitors for MPN—ruxolitinib (Opzelura; Incyte), fedratinib (Inrebic; Bristol Myers Squibb), and pacritinib (Vonjo; CTI BioPharma Corp)—which were approved based on results from the COMFORT-I (NCT00952289), JAKARTA (NCT01437787), and PERSIST-2 (NCT02055781) pivotal trials, respectively.
Ruxolitinib and fedratinib are primarily for patients with intermediate- or high-risk myelofibrosis, including intermediate-2 risk and primary and post-PV/ET myelofibrosis, Mahmoudjafari explained. Pacritinib is indicated for patients with a platelet count below 50,000; all 3 JAK inhibitors have expected adverse event (AE) profiles, which include thrombocytopenia, anemia, bruising, dizziness, headache, and diarrhea.
Although the only true potentially curative treatment for myelofibrosis is transplant, there is a 30% mortality risk associated with it, Mahmoudjafari said. Further, patient adherence remains a predominant issue in patient care, according to Connor Roth, PharmD, BCOP, hematology/oncology pharmacy specialist with Franciscan Alliance, Inc in Chicago, Illinois. Whether due to dosing schedule, toxicities, cost, or all these reasons, people remain forgetful, Roth said. It is much harder to contact patients with a reminder via phone call because “nobody picks up [a call from] a phone number they don’t know,” Roth added.
Tammy McClellan, PharmD, a clinical oncology pharmacist at Riverside Healthcare in Kankakee, Illinois, said one of the greatest unmet needs she is seeing is timely access to medications. The faster a patient can get on a proper treatment regimen, the better they can prevent a blood-clotting event.
Insurance is another barrier to access; however, pharmacists understand how to work within the system and are best positioned to advocate for patients, according to Roth. Location is equally important for access to medications because patients living close to a city can access treatment centers and pharmacies more easily than those in a rural setting. Patients in cities also have better access to clinical trials, Preston said.
McClellan noted that an unmet patient need is effective communication with care providers. She said patients frequently mention that their provider does not listen to their input often enough.
McClellan said a solution may be individualized patient care. Further, Latha Radhakrishnan, PharmD, BCOP, BCPS, a clinical oncology pharmacist and an assistant professor in the College of Pharmacy at the University of Illinois at Chicago, noted that pharmacists and providers can foster improved individualized care through better organized collaboration with the patient and care team. This can make it easier to manage AEs and drug-drug interactions because treatment can be exceedingly difficult, according to Mahmoudjafari. Therefore, improving AE management can improve patient quality of life. “[Symptoms can be] enough to drive these patients absolutely insane,” McClellan added.
Additionally, financial burden is a significant issue for many patients. For this reason, some clinics have financial navigators who work with pharmacists and patients to coordinate benefits, co-pays, and prior authorization. Other institutions may assign these tasks to specialty pharmacists, who typically have experience with patient assistance programs that help older adults or individuals with limited resources to access affordable medications via grants, foundational support, or other means. Ideally, insurance or patient assistance would be connected to the patient’s electronic medical record, according to Roth. The panelists also emphasized patient education. “I really try to explain to [patients], in layman’s terms, what’s going on and just listen to what their issues are,” Preston said.
The panelists said that a best practice is to provide as much information about the disease state and treatment as possible. Because many patients do not understand their disease state, improving their understanding can provide the patient with more control, which can lead them to feeling better able to express concerns and be their own advocate.
“You can’t make the assumption that the patient already knows [everything],” Mahmoudjafari said. This is especially important because oncologists or other providers may be struggling to keep up with a complicated, changing treatment and guidelines landscape.
“Guidelines are dividing, and there’s so many things to know [about the drugs],” Roth said. “Pharmacists can be the ones to extend the hands of the physicians and be a patient advocate when [the patient] doesn’t always have one.”
Reference
American Society for Clinical Oncology. Pharmacy Times Clinical forum. 2023 American Society of Clinical Oncology Annual Meeting; June 2-6, 2023; Chicago, IL. Accessed July 13, 2023. https://conferences.asco.org/am/attend
The FDA granted fast track designation to two oncology therapies.
Selinexor (Karyopharm Therapeutics) — a selective inhibitor of nuclear export — received the designation for treatment of myelofibrosis, including primary myelofibrosis, post-essential thrombocythemia myelofibrosis and post-polycythemia vera myelofibrosis.
“Selinexor’s unique mechanism of action, XPO1 inhibition, is a novel and potentially fundamental mechanism in myelofibrosis,” Reshma Rangwala, MD, PhD, chief medical officer of Karyopharm, said in a company-issued press release.
The company initiated a pivotal phase 3 trial to assess the efficacy and safety of once-weekly selinexor in combination with ruxolitinib (Jakafi, Incyte) for JAK-naive patients with myelofibrosis. Initial data are expected in 2025.
BRG01 (Biosyngen) — an adoptive immune cell therapy — received the designation for treatment of certain patients with relapsed or metastatic nasopharyngeal carcinoma. The designation applies to use of the agent by patients with Epstein-Barr virus-positive disease.
FDA granted orphan drug designation to BRG01 earlier this year.
References:
Karyopharm receives FDA fast track designation for selinexor for the treatment of myelofibrosis (press release). Available at: https://investors.karyopharm.com/2023-07-17-Karyopharm-Receives-FDA-Fast-Track-Designation-for-Selinexor-for-the-Treatment-of-Myelofibrosis. Published July 17, 2023. Accessed July 19, 2023.
Biosyngen’s first-in-class cell therapy BRG01 receives FDA fast track designation (press release). Available at: https://www.biosyngen.com/index.php?m=home&c=View&a=index&aid=107. Published July 10, 2023. Accessed July 19, 2023.
Haifa Kathrin Al-Ali, MD, provides background on the phase 1/2 study of BMS-986158, presents initial efficacy and safety data from the study, and discusses her hope that novel combination regimens like these could achieve the challenging goal of disease modification in myelofibrosis in the future.
The investigational, oral BET inhibitor BMS-986158 administered with either first-line ruxolitinib (Rituxan) or second-line fedratinib (Inrebic) showcased early efficacy and tolerability in patients with intermediate- or high-risk myelofibrosis. These data suggest that strategies combining BET and JAK inhibition can not only address myelofibrosis-related symptoms but may show potential for disease modification, according to Haifa Kathrin Al-Ali, MD.
Findings from the dose-escalation portion of the phase 1/2 CA011-023 trial (NCT04817007) were reported at the 2023 EHA Congress, and showed that both regimens had manageable toxicity profiles. In part 1A of the study, 82% of patients given BMS-986158 plus ruxolitinib experienced an any-grade treatment-related adverse effect (TRAE); this percentage was 75% in part 1B, which evaluated BMS-986158 plus fedratinib. Dose-limiting toxicities (DLTs) occurred in 2 patients in part 1A and 3 patients in part 1B.
Early efficacy data demonstrated that first-line BMS-986158 plus ruxolitinib led to spleen volume reduction (SVR) that became particularly robust by week 24. By week 48, 80% of patients (95% CI, 28%-100%) given the first-line ruxolitinib combination (n = 5) and 50% of those given the second-line fedratinib regimen experienced an SVR of at least 35%. In the ruxolitinib arm, the mean spleen volume change was –46.7% at week 12, –59.9% at week 24, and –56.3% at week 48; in the fedratinib arm, these percentages were –29.1%, -30.8%, and -33.0%, respectively.
“There is still a way to go, but these preliminary data are quite encouraging,” said Al-Ali, a professor of Translational Oncology and head of the Krukenberg Cancer Center at the University Hospital of Halle (Saale) in Germany.
In an interview with OncLive®, Al-Ali provided background on the phase 1/2 study of BMS-986158, presented initial efficacy and safety data from the study, and discussed her hope that novel combination regimens like these could achieve the challenging goal of disease modification in myelofibrosis in the future.
OncLive: What was the rationale for investigating the use of BET inhibitors as monotherapy or in combination with JAK inhibitors in myelofibrosis?
Al-Ali: We know that in patients [with myelofibrosis] there is a NF-κB–mediated pro-inflammatory cytokine profile. [This] leads to a dysregulated bone marrow microenvironment and osteoblastic differentiation, which contributes to the bone marrow fibrosis. It’s rational to use BET inhibitors because they have been shown to reduce or inhibit the expression of BET-targeted oncogenes like c-MYC and MYC.
Please describe the design of this study. Which patients were included this analysis, and what were the key objectives of the research?
This is an open-label, phase 1b/2 study. It mainly included patients with myelofibrosis who had splenomegaly and [had] either intermediate-1 [disease] plus symptoms, intermediate-2 [disease], or high-risk [disease, according to the Dynamic International Prognostic Scoring System]. The trial consisted of a dose-escalation phase followed by a dose-expansion phase. In the dose-escalation phase, there were also 2 parts. Part 1A [involved] first-line treatment with the BET inhibitor plus ruxolitinib in patients who had no previous exposure to ruxolitinib. [Part 1B consisted of] the second-line combination, [where] the BET inhibitor was combined with 400 mg of fedratinib and [was administered] once daily [to] patients with either intolerance or resistance to ruxolitinib.
This was a phase 1 study. [At the 2023 EHA Congress,] we presented the data from the dose-escalation phase, so the primary objective is always safety. The secondary objective was efficacy in terms of SVR. There were some exploratory analyses on JAK2 allele burden as well as bone marrow fibrosis.
According to data presented at the congress, what should be known about the safety of BMS-986158?
Regarding safety, [we] found that both the first-line combination with ruxolitinib or the second-line combination with fedratinib were feasible, tolerable, and the safety profiles were manageable. The main AE was thrombocytopenia, which is a class effect; it’s manageable and transient with dose modification or dose holding. The second major AEs were gastrointestinal [toxicities, including] diarrhea and nausea. Generally, these [effects] were mild, never led to the discontinuation of patients from the trial and were quite manageable.
What were the efficacy findings reported with these combinations?
Regarding efficacy, there are very promising results [showing] SVR of at least 35% from baseline by MRI. In the first-line cohort, there was a rapid and relevant reduction from baseline spleen volume [of at least 35%] in [73%] of patients [at week 12]. This seemed to be sustainable. Looking at SVR at week 24, 100% of patients [experienced] SVR. This is a phase 1 [study], and we should be careful, but these are encouraging results.
[Similar results were seen] with the second-line treatment, although the [duration of] follow-up was shorter. At week 12, at least [38%] of the patients [experienced] more than a 35% reduction in spleen volume. These are also encouraging results.
Finally, evidence for disease modification might be seen regarding JAK2 allele burden reduction. [This was] seen quite early, starting by cycle 6 in all the patients [with] JAK2-mutated [disease]. Additionally, in patients with follow-up bone marrow biopsies that could be evaluated, there seems to be a significant reduction in at least 1 grade of fibrosis by week 12 or week 24. [The study includes a] small number of patients, and these are preliminary, encouraging data. This bone marrow fibrosis regression seems to be associated with a hematological, [specifically] anemic, response.
You mentioned that potential evidence for disease modification may have been observed with this in the form of JAK2 allele burden reduction. In myelofibrosis, what efforts are currently underway to develop disease-modifying therapies that go beyond standard approaches focused on symptom management?
One of the major challenges [in myelofibrosis] is to see [clear evidence of successful] disease modification. All the biomarkers you can measure, like bone marrow fibrosis or a reduction in allele mutational burden, should have a clinical outcome correlation. This is a big challenge.
In the future, it is crucial to move away from only SVR and symptom improvement. We have great drugs that could do that. We have to wait for data from phase 3 randomized trials, and we need time to learn and [understand] the benefit of these combination treatments. My wish is to [achieve] sustainable, durable, clinical responses for patients with these combinations, but this is still an area with a lot of unanswered questions.
Are any next steps planned for the investigation of BMS-986158 in this disease?
The next step [for this research] is going further with the trial. The expansion phase has started for the first-line combination treatment in patients who are ruxolitinib naïve. The same will hopefully be happening for the second-line treatment. If these all [show] positive signals, we will move to a phase 3 clinical trial.
What is your main takeaway message for colleagues regarding this presentation?
The takeaway message is that it is feasible to have these combinations. We have learned how to dose [them] and how to manage AEs, which is the first step [for] every treatment. We have [also] shown encouraging preliminary clinical data regarding SVR, [as well as] some encouraging translational aspects like reduction in JAK allele burden or improvement in bone marrow fibrosis.
Overall, what was most exciting about this year’s EHA Congress?
[This year’s EHA Congress has] been particularly exciting. It is almost [solely] a myeloproliferative neoplasm congress because [there are] so many new, exciting datasets [being presented]—not only for a myelofibrosis, but also for polycythemia vera and essential thrombocythemia. At the end of the day, the most important winners will be the patients.
Reference
Ayala R, Lopez N, Abulafia AS, et al. BMS-986158, a potent BET inhibitor, as monotherapy and in combination with ruxolitinib or fedratinib in intermediate- or high-risk myelofibrosis (MF): results from a phase 1/2 study. Presented at the 2023 EHA Congress; June 8-11, 2023; Frankfurt, Germany. Abstract S213.
Analysis published in the Journal of Comparative Effectiveness Research shows ropeginterferon alfa-2b-njft provided a cost-effective benefit in quality-adjusted life years compared to a commonly used treatment pathway
BURLINGTON, Mass.–(BUSINESS WIRE)– PharmaEssentia USA Corporation, a subsidiary of PharmaEssentia Corporation (TPEx:6446), a global biopharmaceutical innovator based in Taiwan leveraging deep expertise and proven scientific principles to deliver new biologics in hematology and oncology, today announced the publication of a cost-effectiveness analysis of ropeginterferon alfa-2b-njft (marketed as BESREMi®) in the Journal of Comparative Effectiveness Research. The analysis, titled “Cost-Effectiveness of Ropeginterferon Alfa-2b-njft for the Treatment of Polycythemia Vera,” showed that ropeginterferon alfa-2b-njft provided a cost-effective benefit for a broad range of patients with polycythemia vera (PV) versus first-line hydroxyurea followed by ruxolitinib. Cost effectiveness was demonstrated in a modeled population including both low- and high-risk patients receiving first- or second-line treatment with ropeginterferon alfa-2b-njft.
“PV is a chronic blood cancer that requires lifelong therapy, which can prevent or delay negative clinical outcomes, but is also associated with additional costs to patients and payers. These new data demonstrate ropeginterferon alfa-2b-njft’s value not only to people living with PV, but also to the healthcare system at large,” said Dr. Aaron Gerds, MD, MS, Medical Director at Case Comprehensive Cancer Center and lead study author. “This study shows ropeginterferon alfa-2b-njft is a cost-effective treatment option over the modeled lifetime and that earlier initiation of treatment of PV with effective therapy can translate to more favorable cost to benefit ratios.”
PV is the most common myeloproliferative neoplasm (MPN) and a long-term, potentially life-threatening disease with limited approved treatment options. Patients with PV are at risk for disease progression to myelofibrosis (post-PV MF) or transformation to blast phase (MPN-BP) which is akin to acute myeloid leukemia1-3. Patients are also at an increased risk for arterial and venous thromboembolic events (TEs) which are associated with higher mortality, lower quality of life, and higher healthcare costs4-7.
This new study used an economic model developed from the United States healthcare system perspective. Inputs were informed by the data from randomized clinical trials, including the PROUD-PV and CONTINUATION-PV studies, and from real-world sources. The model compared ropeginterferon alfa-2b-njft used either as first- or second-line therapy versus an alternative treatment pathway of first-line hydroxyurea followed by ruxolitinib. To reflect the long-term consequences of treating PV, results were presented over a lifetime horizon.
Findings from the study conclude ropeginterferon alfa-2b-njft is a cost-effective treatment option for a broad range of patients with PV, including both low- and high-risk patients and patients with and without prior cytoreductive treatment with hydroxyurea. Of note:
These data show that over the modeled lifetime, patients who receive ropeginterferon alfa-2b-njft have more years alive (0.4), higher quality-adjusted life years (QALYs) (0.4), and higher cost ($60,175) as compared to the alternative treatment pathway. Weighing the additional costs versus the additional QALY gains results in a cost per QALY of $141,783.
This cost per QALY is less than a standard willingness to pay threshold of $150,000 per QALY8.
In this study, treating patients at a younger age or those with low-risk disease led to more cost-effective results, suggesting that earlier initiation of treatment of PV with effective therapy can translate to more favorable cost to benefit ratios.
The model was sensitive to treatment costs, the percentage of patients who discontinue hydroxyurea, the percentage of ropeginterferon alfa-2b-njft users who switch to monthly dosing, the percentage of ropeginterferon alfa-2b-njft users as 2nd line treatment, and the treatment response rates.
“Ropeginterferon alfa-2b-njft has demonstrated safety and efficacy in studies including both low- and high-risk patients and patients with and without prior cytoreductive treatment with HU. Recently, the National Comprehensive Cancer Network (NCCN) updated their treatment guidelines to include ropeginterferon as a preferred treatment regimen for both low- and high-risk PV patients9. As ropeginterferon continues to be more widely used in clinical practice, this study was imperative to demonstrate the cost-effectiveness of ropeginterferon alfa-2b-njft for a broad range of patients with PV,” said Raymond Urbanski, M.D., Ph.D., Senior Vice President and U.S. Head of Clinical Development and Medical Affairs at PharmaEssentia. “This analysis also underscores the importance of treating low-risk patients and patients early in their disease journey to help minimize more severe events and their corresponding costs to the healthcare system.”
The full benefits of ropeginterferon alfa-2b-njft may not be fully captured in the model, and in particular, data on disutility of phlebotomy are lacking. Although some patients may tolerate regular phlebotomies, others can experience iron deficiency which can negatively impact quality of life. While the results from a scenario analysis incorporating a small decrement in utility had minimal impact on the cost-effectiveness results, due to the lack of data in this area, this estimate remains conservative. This study took a U.S. healthcare perspective, and the results may not generalize to other countries given the differences in healthcare resource use, costs and cost-effectiveness thresholds.
About Polycythemia Vera (PV)
Polycythemia vera (PV) is a cancer originating from a disease-initiating stem cell in the bone marrow resulting in a chronic increase of red blood cells, white blood cells, and platelets. PV may result in cardiovascular complications such as thrombosis and embolism, and often transforms to secondary myelofibrosis or leukemia. While the molecular mechanism underlying PV is still subject of intense research, current results point to a set of acquired mutations, the most important being a mutant form of JAK2.10
About BESREMi® (ropeginterferon alfa-2b-njft)
BESREMi is an innovative monopegylated, long-acting interferon. With its unique pegylation technology, BESREMi has a long duration of activity in the body and is aimed to be administered once every two weeks (or every four weeks with hematological stability for at least one year), allowing flexible dosing that helps meet the individual needs of patients.
BESREMi has orphan drug designation for the treatment of polycythemia vera (PV) in adults in the United States. The product was approved by the European Medicines Agency (EMA) in 2019, in the United States in 2021, and has recently received approval in Taiwan and South Korea. The drug candidate was invented by PharmaEssentia and is manufactured in the company’s Taichung plant, which was cGMP certified by TFDA in 2017 and by EMA in January 2018. PharmaEssentia retains full global intellectual property rights for the product in all indications.
Patients with previously treated myelofibrosis (a type of myeloproliferative neoplasm) tended to benefit more regarding symptom, spleen and anemia outcomes when administered a regimen of momelotinib compared with those given Danocrine (danazol), according to updated findings of the recently completed phase 3 MOMENTUM trial.
“Momelotinib was associated with durable symptom, spleen, and anemia benefits, late responses after week 24 and favorable safety through week 48. These results highlight the potential benefits of treatment with momelotinib in patients with myelofibrosis, particularly those with anemia,” the researchers wrote in their findings, which were published in the journal The Lancet Hematology.
The study involved 195 adults with post-polycythemia vera or post-essential thrombocythemia myelofibrosis that were previously treated with a JAK inhibitor for 90 days or more. Two-thirds of participants (130 patients) were randomly assigned to receive momelotinib, while the other third (65 patients) were randomly assigned to receive Danocrine.
After 24 weeks, all patients were eligible to receive momelotinib, and 93 (72%) and 41 (63%) in the momelotinib and Danocrine groups, respectively, entered the momelotinib open-label extension period.
Among the patients who continued on momelotinib treatment and were evaluable based on total symptom score criteria, 30 (45%) patients in the original momelotinib group and 15 (50%) in the Danocrine groups responded to treatment, meaning that their disease shrank or disappeared from the therapy.
A total of 45 patients (34.62%) in the momelotinib group died of any cause compared with 26 (40%) in the Danocrine group.
Momelotinib works by inhibiting JAK2 signaling pathway. Of note, JAK2 is a genetic mutation that is commonly found in patients with myeloproliferative neoplasms and leads to scarring in the bone marrow. This scarring inhibits the marrow’s ability to produce healthy blood cells — a condition known as myelofibrosis.
Long-term follow-up revealed no new side effects from momelotinib, with the most common non-blood-related side effects from the drug being diarrhea (45 patients [26%] in the momelotinib group) and weakness or lack of energy (28 [16%]). The most common moderate to severe (grade 3 or 4) side effects were thrombocytopenia (33 [19%]) and anemia (30 [18%]). A total of 79 patients (46%) given momelotinib experienced at least one serious side effect, and 30 patients (18%) died from a treatment-emergent side effect, with two fatal treatment-emergent side effects considered to be potentially linked to momelotinib.
Tuba Ersal, Vildan Özkocaman, İbrahim Ethem Pınar, Cumali Yalçın, Bedrettin Orhan, Ömer Candar, Sinem Çubukçu, Tuba Güllü Koca, Fazıl Çağrı Hunutlu, Şeyma Yavuz, Rıdvan Ali & Fahir Özkalemkaş
The impact of inflammatory markers such as systemic immune-inflammation (SII) index and systemic inflammation response index (SIRI) on myelofibrosis (MF) prognosis was evaluated for the first time in this study. Data from 60 patients diagnosed with MF between March 2011 and September 2022 were retrospectively analyzed. In addition to disease-related markers, the impact of SII and SIRI on prognosis was evaluated. In our study, the overall median survival (OS) was 64 months. OS was significantly shorter in patients older than 65 years, with high ferritin and lymphocyte levels, transfusion dependence at diagnosis, platelet count below 100 × 109/L, Hb level below 8 g/dl, and high risk according to the dynamic international prognostic scoring system (DIPSS)-Plus score. When these variables were included in the multivariate Cox regression model, it was found that being older than 65 years, having a high ferritin value, being at high risk according to the DIPSS-plus score and Hb values below 8 increased the risk of death. Platelet-to-lymphocyte ratio (PLR) and SII index were lower in patients with a fatal outcome. No statistically significant relationship was found between SIRI and mortality. The findings of this study showed that low PLR and high ferritin were associated with poor prognosis in MF. Elevated SII and SIRI, evaluated for the first time in patients with myelofibrosis, did not predict prognosis. Since non-inflammatory variables play a role in the pathogenesis of MF, bone marrow indicators and systemic inflammation indicators derived from hematologic parameters may not be accurate.
Introduction
Myelofibrosis (MF) is a BCR-ABL1-negative myeloproliferative neoplasms (MPN) characterized by anemia, extramedullary hematopoiesis, bone marrow fibrosis, splenomegaly, constitutional symptoms, and acute myeloid leukemia progression1. Most patients carry a mutation in the JAK-2, CALR, or MPL genes2, which contributes to the JAK-2-signal-transducer-and-activator-of-transcription signaling pathway and the high inflammatory state characteristic of these diseases. Inflammation plays a crucial role in the development and progression of MPN.
The prognosis of MF varies greatly. While some patients only have a few months to survive, others live for more than 20 years. The three leading causes of death are hemorrhage, infection brought on by bone marrow loss, and transformation into acute leukemia3.
A good risk stratification model provides information about the prognosis of patients, affecting the decision whether the patient is included as a candidate for allogeneic stem cell transplantation and thereby the treatment. To evaluate the mortality risk of MF patients, the dynamic international prognostic scoring system (DIPSS)4 or DIPSS-plus is generally used5.
It is well established that inflammation affects all phases of tumor growth and can increase the risk of developing a tumor (triggering the first genetic mutation, tumor development, metastasis, and progression). Therefore, inflammation parameters are strong candidates for predicting cancer prognosis. Numerous inflammatory indicators have recently been linked to a poor prognosis for cancer, including C-reaction protein (CRP), neutrophil-to-lymphocyte ratio (NLR), and platelet-to-lymphocyte ratio (PLR)6,7,8. The systemic immune-inflammation (SII), which was first used in hepatocellular cancer in 2014 as a new inflammation marker based on peripheral neutrophil, platelet, and lymphocyte counts9 and the systemic inflammation response index (SIRI), which was developed to predict survival in patients with pancreatic cancer and based on peripheral neutrophil, monocyte, and lymphocyte counts10, have also been used as inflammatory biomarkers to predict prognosis in many cancer types11,12,13,14.
In this study, in addition to the potential prognostic markers examined in other retrospective studies to date, we investigated the prognostic value of SII and SIRI in MF for the first time in the literature.
Materials and methods
The data of 60 patients who were followed up with the diagnosis of MF between March 2011 and September 2022 at Bursa Uludag University Hematology Department Clinic were retrospectively analyzed. All patients met the 2016 World Health Organization criteria for PMF15 or the 2008 international working group for myelofibrosis research and treatment (IWG-MRT) criteria for SMF16. Patients were divided into four groups as low, intermediate-1, intermediate-2, and high-risk groups according to DIPSS and DIPSS-plus scores. The effect of age, MF subtype, JAK-2 mutation status, erythrocyte transfusion dependence, presence of constitutional symptoms, splenomegaly, leukocyte, hemoglobin, platelet, CRP, mean corpuscular volume, mean platelet volume (MPV), red blood cell distribution width (RDW), LDH, ferritin levels, TS and CAR, PNI, NLR, PLR, leukocyte to lymphocyte ratio (WLR), ferritin to lymphocyte ratio (FLR), lymphocyte to LDH ratio (LLR), DIPSS and DIPSS-plus risk group, and SII and SIRI, which were examined for the first time in MF, on prognosis was investigated in all patients. SII was calculated as platelet count × neutrophil count/lymphocyte count in peripheral blood and SIRI was calculated as neutrophil count × monocyte count/lymphocyte count. Mortality was defined as patients who died during follow-up. Our study was conducted under the institutional research committee’s ethical standards and according to the 1964 Helsinki Declaration. This study was approved by the clinical research ethics committee of Bursa Uludag University Faculty of Medicine (Decision No: 2022-18/20).
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics for Windows Version 25.0 (Statistical Package for the Social Sciences, IBM Corp., Armonk, NY, USA). Descriptive statistics were presented as n and % for categorical variables and mean ± SD, median (IQR) for continuous variables. Data were analyzed in terms of normality assumptions. For continuous variables with Kolmogorov–Smirnov values p > 0.05, independent samples t-test was used as the parametric test to evaluate the difference in mortality between the groups. Chi-square test was used to compare categorical variables. Three receiver operating characteristic (ROC) curve analysis was performed for SII and ferritin values to predict mortality. Kaplan–Meier method was used to compare survival times between various variables. Finally, multivariate Cox regression results of various clinical factors on mortality risk were presented. p < 0.05 was considered statistically significant in all analyses.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent: Informed consent was obtained from all individual participants included in the study.
Results
Sixty patients made up the study population, with 53.3% of the females and 46.7% of the males. Median age at diagnosis of 63 years. Most patients (76.6%) and those with splenomegaly (93.3%) had anemia at the time of diagnosis. Of these patients, 41.6% had massive splenomegaly. There were 41.6% of cases of constitutional symptoms, with weight loss being the most prevalent (61.5%) and high fever being the least prevalent (11.5%). Thrombosis was present in 16.6% of patients (n = 10). Four patients had cerebral vascular thrombosis, five had portal system thrombosis (three portal vein thrombosis, two splenic infarction), and one had deep vein thrombosis. JAK-2 mutation positivity was detected in 30 (58.8%) of 51 patients who underwent genetic screening. The rate of transformation to acute leukemia was 8.3%. Table 1 shows the distribution of the demographic and clinical findings of the patients.
Table 1 Demographic and clinical characteristics of the patients.
As seen in Table 2, median overall median survival (OS) was 64 months (95% CI 54.88–73.10). Two-year OS was 73.6%, while five-year OS was 55.6%. No difference was found between the MF subtypes (p = 0.825). Median OS was significantly different between the age groups (p = 0.005) (Fig. 1). Median survival was 73.7 months in patients under 65 years of age (95% CI 68.07–79.32) compared to 44.6 months in patients over 65 years of age (95% CI 10.66–78.53). Two-year OS and five-year OS were 83.3% and 74.1% in patients under 65 years of age compared with 56.4% and 25.6% in patients over 65 years of age.
Table 2 OS comparisons according to patient variables.
A significant difference was found in OS with respect to DIPSS-plus risk groups (p < 0.001). Median survival was 73.7 months (95% CI 47.43–99.66) in the low-risk group, 72.1 months (95% CI –) in the intermediate-1 risk group, 59.7 months (95% CI 31.17–82.22) in the intermediate-2 risk group, and 9.6 months in the high-risk group (95% CI 8.06–11.4). A statistically significant difference was also found in median OS between the high-risk group and all other risk groups (p < 0.001). Two-year OS and five-year OS were 100% and 83.3% in the low-risk group, compared with 92.3% and 84.6% in the intermediate-1 risk group. In the intermediate-2 risk group, two-year OS was 76.3% and five-year OS was 49.9%. In the high-risk group, all patients died within two years.
PLR (p = 0.048), SII (p = 0.018) and lymphocyte count (p = 0.033) showed a statistically significant difference between patients with and without mortality. PLR and SII were lower in patients with mortality compared to patients without mortality, while ferritin and lymphocyte levels were higher. No significant difference was found in survival with respect to other variables (neutrophils, platelets, NLR, LLR, WLR, FLR, RDW, MPV, CRP, LDH, spleen size, serum iron, iron binding capacity, TS, PNI, CAR).
A significant relationship was found between OS and Hb levels below 8 g/dL (p = 0.027), transfusion dependency (p < 0.001), and platelet count below 100 × 109/L (p = 0.002), while no statistically significant relationship was found between OS and MF subtypes, positive or negative JAK-2 mutation status, presence of constitutional symptoms, TS < 20%, Hb < 10 g/dL, leukocyte count > 25 × 109/L, degree of collagen and reticulin fibrosis, and DIPSS risk groups.
Univariate analysis results showed that age, ferritin, transfusion dependency, platelet count, DIPSS-plus risk group, Hb, and SII index variables were statistically significant for mortality risk (p < 0.05). The variables that were significant in univariate analyses were included in the multivariate Cox regression model. According to the results of the multivariate Cox regression model, it was found that being over 65 years of age (HR 7.29; 95% CI 2.44–21.75; p < 0.001), increased ferritin values (HR 1.00; 95% CI 1.00–1.01 p = 0.002), high-risk DIPSS-plus (HR 12.63; 95% CI 1.30–122.30 p = 0.029), and hemoglobin values below 8 increased mortality risk (OR 0.32; 95% CI 0.11–0.94 p = 0.038) (p < 0.001, − 2 loglikelihood = 158,326) (Table 3).
Table 3 Multivariate Cox regression results for various clinical variables.
The predictive power of SII for mortality was statistically significant (p = 0.032). In the ROC analysis conducted to evaluate SII in predicting mortality, the area under the curve was 0.677 (95% CI 0.528–0.827) and the cut-off value for SII was 1246.78 (Fig. 2). For this value and below, sensitivity was 57.6% and specificity was 55.5%. The predictive power of ferritin was not statistically significant for mortality (p = 0.097) (Table 4, Fig. 3).
Accurate risk assessment is crucial for developing the best treatment strategy in MF, which is one of the BCR-ABL-negative MPNs accepted as a model of inflammation-related cancer development, especially in young patients. Widely accepted scoring systems require genetic evaluation and tests may be difficult to access in some centers. The current study assessed prognostic indicators in patients with MF. To the best of our knowledge, this is the first study to have examined the relationship between SII and SIRI and mortality in MF patients in the literature. Inflammatory indicators and parameters that could affect prognosis were assessed in 60 patients.
Indices such as SII and SIRI are thought to be associated with the prognosis of various tumors. A meta-analysis by Yang et al. evaluating 22 studies including 7657 patients revealed that high SII was clearly associated with lower OS, time to recurrence, progression-free survival, cancer-specific survival, relapse-free survival, and disease-free survival17. These results suggest that high SII may be a potential prognostic marker in patients with various cancers and may be associated with poor overall outcomes. A study by Geng et al. in patients with esophageal cancer showed that median OS was significantly higher in patients with low SIRI18.
In this study, the relationship between MF and SII was evaluated for the first time in the literature; and paradoxically, mortality was found to be lower in patients with MF compared to patients without MF (p = 0.018). This discrepancy results from patients with a fatal course having higher lymphocyte numbers and lower platelet counts. SII lost its relevance when these variables were incorporated into the multivariate Cox regression model. Additionally, SIRI was also examined for the first time in MF patients and found not to be associated with mortality (p = 0.492).
Anemia is a disease characteristic most consistently associated with poor prognosis in MF5,19,20,21. The most commonly used threshold in prognostic models is 10 g/dL. Transfusion dependence has had poor prognostic significance in MF22,23,24. There is ongoing debate about the relationship between transfusion dependence and poor prognosis in chronic MPN. Some authors argue that transfusion dependence affects survival through the adverse effects of chronic erythrocyte transfusion, such as iron overload and transfusion-related immunomodulation. In the present study, when the Hb cut-off point was taken as 10 g/dL, no difference was found between the groups in terms of survival (p = 0.168). However, when the cut-off point was taken as 8 g/dL, a significant difference was found in median OS (p < 0.027). Hb level ≤ 8 g/dL was determined as a marker of poor prognosis. When included in the multivariate Cox regression model, Hb < 8 g/dL increases the risk of mortality (HR 0.32; 95% CI 0.11–0.94 p = 0.038).
Some studies have found that thrombocytopenia was associated with poor prognosis5,20,21,25, but it was noted that low platelet counts are frequently associated with anemia and collinearity in multivariate regression models may make it difficult to characterize thrombocytopenia as an independent prognostic factor19. In the present study, platelet count was lower in patients with a mortal course, but the difference was not statistically significant (p = 0.085). When the cut-off value for platelet count was taken as 100 × 109/L, a significant difference was found in median OS (p = 0.002). Median OS was significantly shorter in patients with platelet count below 100 × 109/L (72.1 months versus 17.1 months).
The transfusion dependency at diagnosis or during MF is an indicator of poor prognosis22,23. In our patients, median OS was 73.7 months in the group without transfusion dependence and 17.1 months in the group with transfusion dependence, which was significantly lower (p < 0.001).
Consistent with previous studies, age was associated with OS in both univariate analysis and multivariate Cox regression analysis. When the cut-off point for age was taken as 65 years, a significant difference was found in OS between the groups (p = 0.005). When included in the multivariate Cox regression model, it was found that mortality risk was significantly higher in those older than 65 years (HR 7.29; 95% CI 2.44–21.75; p < 0.001).
Currently, the most widely used prognostic scoring system in MF is DIPSS-plus. DIPSS-plus was used in 967 consecutive patients at the Mayo Clinic and resulted in median survival of 1.8, 3.6, 7.8, and 17.5 years for high, intermediate-2, intermediate-1, and low-risk patients, respectively26. When the patients in this study were divided into risk groups according to DIPSS-plus, a statistically significant difference was found between the median OS times (p < 0.001). Median OS was 73.7 months in the low-risk group, 72.1 months in the intermediate-1 risk group, 59.7 months in the intermediate-2 risk group, whereas it was 9.6 months in the high-risk group (p < 0.001). In the multivariate Cox regression analysis, a significant difference was found between the low-risk group and the high-risk group persisted (HR 12.63; 95% CI 1.30–122.30 p = 0.029), while the significance between the low-risk group and intermediate-1 and intermediate-2 risk groups disappeared (p = 0.151, p = 0.570, respectively).
With respect to iron metabolism, studies have shown that high ferritin value and low TS are associated with low OS in MF. Lucijanic et al. evaluated the prognostic impact of low TS in 87 patients with PMF. Low TS was found to have a detrimental effect on the survival of PMF patients, independent of anemia and ferritin levels27. In the present study, ferritin level was found to be higher in patients with mortality (p = 0.024) and when included in the multivariate Cox regression model, it was found that an increase in ferritin levels increased the risk of mortality (HR 1.00; 95% CI 1.00–1.01 p = 0.002). However, there was no statistically significant relationship between TS, serum iron level, iron binding capacity, RDW, and mortality. Numerous studies have been published in the literature showing the link between several inflammation markers, including NLR and PLR, and a poor prognosis for cancer7,8,28. In a study evaluating NLR and PLR in MF, these values were found to be significantly higher in patients compared to healthy controls. In univariate analyses, shorter overall survival was observed in patients presenting with high NLR and low PLR29. In the same study, increased RDW was associated with survival (p = 0.039). In MF, high CRP is associated with features of more advanced disease and a trend toward worse clinical outcomes as part of individual parameters or different prognostic scores30,31,32. In this study, no correlation was found between CRP and NLR and survival. Consistent with the literature, survival was shorter in patients with low PLR values (p = 0.048).
CAR has recently been recognized as an inflammatory biomarker and prognostic factor in several malignant neoplasms33,34. However, a study evaluating CAR in patients with MF reported that higher CAR was associated with lower OS35. PNI is an index reflecting a patient’s inflammatory, nutritional, and immune status. In a study evaluating PNI in MF patients, low PNI predicted worse survival independent of DIPSS36. In the present study, however, CAR and PNI had no effect on survival.
The mean LDH level was higher (p = 0.108) and spleen size was larger (p = 0.122) in patients with a mortal outcome, but this was not statistically significant.
This study has certain limitations. The study was conducted retrospectively and in a single-center.
In conclusion, the results obtained in this study show that elevated SII and SIRI, which have prognostic significance for many cancers, cannot be used as markers for poor prognosis in MF. Since the pathology of MF directly involves the bone marrow unlike solid organ cancers, these inflammation markers may be insufficient to predict prognosis. Further clinical studies are needed to confirm these results.
Alessandra Carabbio, Alessandro Maria Vannucchi, Elisa Rumi, Valerio De Stefano, Alessandro Rambaldi, Giuseppe Carli, Heinz Gisslinger, Francesco Passamonti, Juergen Thiele, Naseema Gangat, and Tiziano Barbui
Ample evidence has been provided that accurate discrimination between essential thrombocythemia (ET) and early prefibrotic primary myelofibrosis (pre-PMF) has an impact not only on presenting laboratory data but also on complications, like thrombosis, progression to overt myelofibrosis (MF), transformation to blast phase (BP), and overall survival [1,2,3,4,5,6,7,8]. However, studies estimating the epidemiology of these critical events in the two entities have mainly focused on one isolated outcome at a time, without considering the entire spectrum of multiple intermediate disease states, possibly affecting probabilities and risk factors of the outcome of interest. This situation calls for a multistate model approach, a technique that allows a more in-depth insight into intermediate factors likely influencing the progressive transitioning from one status to another.
The aim of the present investigation was to estimate the probabilities that intermediate-state passages, including thrombosis, overt MF, and BP, impact the final absorbing state (death) in ET versus pre-PMF. To this purpose, retrospective data from two multicenter and well-documented studies [1, 9] were used: (i) ET patients (n = 791) from a multicenter international study of 891 cases, selected for the availability of complete disease history [1] and (ii) pre-PMF patients (n = 382) from four different Italian centers [9]. Both studies were approved by all institutional review boards or ethical committees of participating centers.
At the time of diagnosis, treatment-naïve ET and pre-PMF patients revealed different hematologic and clinical characteristics (Table S1). A parametric Markov multistate model [10] was applied to analyze data on survival considering intermediate states that are part of the natural history of ET and pre-PMF. The model included five states with ten possible transitions (Fig. S1): all patients begin in the initial state of diagnosis (ET, panel A, n = 791 or pre-PMF, panel B, n = 382) and then they could transit through the occurrence of an incident thrombotic event (Table S2) and/or the evolution to overt MF and/or BP (transient states) before death (absorbing state).
In ET, transition-1 from diagnosis to thrombosis included 101/791 patients (12.7%), but this status was transient in 21/101 patients that moved to death (21%) after a median time of 4.0 years (IQR: 1.6–6.4), 3/101 (6%) and in 1/101 (2%) to MF and BP, after a median time of 4.7 and 5.2 years, respectively. Remarkable was that in pre-PMF, the direct transition to thrombosis was found in 13.9%, a figure not different from ET (i.e., 12.7%). Conversely, pre-PMF substantially differed from ET for a higher rate of direct transition to overt MF or BP, that was 13 and 4% vs. 4 and 0.6%, respectively.
After 10 years, the state occupation probability of being event-free was 70 and 50% in ET and pre-PMF, respectively, and progressively decreased, particularly in pre-PMF (Fig. S2), due to earlier mortality, particularly for a greater probability of hematological evolutions. This trend was even more evident for death; regardless of the pathways through hematological evolutions, deaths were double in pre-PMF than ET, reaching 30, 60, and 80% vs. 15, 30, and 60% at 5, 10, and 20 years, respectively.
Probabilities to direct transition to thrombosis (n = 101 in ET and n = 53 in pre-PMF) and overt MF (n = 29 in ET and n = 51 in pre-PMF) are compared in Fig. 1. The trend of experiencing thrombosis directly after ET diagnosis showed to increase in the first 10 years (10%) and to decline subsequently (less than 5% at 30 years). On the contrary, in the first decade after diagnosis (<5%), the same probability grew slowly in pre-PMF while subsequently rose up to crossing the ET trend (8% after 30 years). Instead, the direct transitions from pre-PMF to overt-MF had an opposite trend: in the first 10 years, it reached a peak of 11%, while in ET, the trend was less pronounced, reaching a probability not exceeding 2.3% in the same period post-diagnosis.
Fig. 1: Direct transition probabilities to thrombosis and evolution in overt MF.
Direct transition probabilities over time from diagnosis of ET or pre-PMF to thrombosis (A) and overt MF (B). Transition probabilities are defined as the probability of going from a given state to the next state in a Markov process. Direct transitions refer to all the 791 and 382 ET and pre-PMF patients, respectively, initially at risk; thus, they represent the probability that a patient can first experience thrombosis or evolve into overt MF.
The performance of the IPSET-thrombosis score [11] was tested in both ET and pre-PMF for the direct transition to thrombosis. In ET, considering the low-risk group as a reference, the intermediate and high-risk groups determined by IPSET-thrombosis were confirmed to predict the thrombotic risk (HR = 2.08, 95% CI = 1.28–3.37, p = 0.003 and HR = 3.13, 95% CI = 1.82–5.40, p < 0.001, respectively). In pre-PMF, the same model was unpowered to reach statistical significance in the intermediate-risk group (HR = 2.50, 95% CI = 0.87–7.21, p = 0.089), while it was in the high-risk category (HR = 3.93, 95% CI = 1.52–10.11, p = 0.005).
Concerning survival, most of the deaths in ET and pre-PMF occurred directly from diagnosis (Fig. 2). The intermediate events that most influenced death were thrombosis (25.3%) in ET and BP (23.8%) in pre-PMF. In comparison with ET, the probability of direct transition from diagnosis to death in patients with pre-PMF increased linearly over time (Fig. 2) and was twofold higher, reaching values of 15, 30, and 60% at 5, 10, and 20 years, respectively. Of note, the probability of death in ET patients with an intermediate thrombosis state maintained a fourfold higher value over time than the ones without thrombosis. As expected, the probabilities of death in MF or BP status were higher and occurred faster, and not substantially different in ET or pre-PMF.
Fig. 2: Transition probabilities to death in ET and pre-PMF.
Comparison of the direct and indirect (via thrombosis, evolution in MF or BP) transition probabilities to death (absorbing state) over time from diagnosis of ET (dash lines) or pre-PMF (solid lines).
We confirmed the good performance of the IPSET-survival score [12] to differentiate the risk of direct mortality in ET (intermediate: HR = 4.38, 95% CI = 1.63–11.73, p = 0.003 and high-risk: HR = 20.17, 95% CI = 7.71–52.78, p < 0.001, compared to low-risk). The IPSET-survival score was equally well performing in pre-PMF (HR = 3.52, 95% CI = 1.22–10.12, p = 0.019 and HR = 13.37, 95% CI = 4.63–38.57, p < 0.001, for intermediate and high-risk groups, respectively, compared to low-risk). However, the discriminatory power of the IPSET-survival in ET was lower when the multistate model evaluated the mortality mediated by the thrombosis state; only high-risk patients were discriminated (HR = 19.27, 95% CI = 2.46–51.01, p = 0.005), whereas the intermediate-risk group was not significantly different from the low-risk one (HR = 3.84, 95% CI = 0.55–26.12, p = 0.194). Thus, in addition to the IPSET-survival risk factors (i.e., age ≥60 years, previous thrombosis, and white blood cells count ≥11 × 109/L) we found that platelets count ≥1000 × 109/L (HR = 5.74, 95% CI = 1.79–18.40, p = 0.003) and arterial vs. venous thrombosis in the follow-up (HR = 4.43, 95% CI = 1.04–18.91, p = 0.044) were independent predictors. The low number of deaths after thrombosis (12/105, 11%) in pre-PMF did not allow us to analyze the IPSET-survival performance in this transition.
The present multistate analysis, provides new insights for a better understanding of ET and pre-PMF disease processes. For example, in pre-PMF, the probability of thrombosis in the first decade was lower (<5%) due to a strong competitor represented by the evolution in MF (up to 11%). Consequently, occupation of thrombosis state in the first decade was lower in pre-PMF than in ET patients but became comparable in the last decades of observation (13 and 14% in ET and pre-PMF, respectively), supporting our previous cumulative estimates obtained with conventional methodology [1]. This notion might have practical implications to differentiate treatments during the course of the two entities by preferring antithrombotic prophylaxis according to IPSET thrombosis in ET that kept its discriminatory power also in this multiple competing adjustment analysis. In pre-PMF, therapy of first choice might be directed to prevent myelofibrosis evolution, provided agents able to do that are positively evaluated in appropriate clinical trials.
Regarding BP evolution, we highlight that the direct transition from the diagnosis was predominant in ET (n = 6/7, 86%), and it was modestly influenced by the pathway through thrombosis (n = 1/7, 14%). Unfortunately, we could not provide sufficient information on the role of cytoreductive therapy in these transitions due to the unreliable timing of drug administration.
Mortality prediction in ET was the topic addressed in a previous study [12]. On the basis of the hazard ratio estimates from Cox regression models, the IPSET-survival model was constructed, and 867 ET patients were allocated into three risk categories with significantly different survival [12]. In the present analysis, in a selected group of patients (n = 791) from the same database, we re-evaluated the risk factors of death considering the possible influence of the intermediate states that occurred before death, and confirmed the performance of the IPSET-survival scoring system for the prediction of direct mortality. However, we also found that the effect on mortality exerted by the intermediate thrombosis state was not negligible (accounting for 25% of deaths) and fourfold higher than in patients without incident thrombosis. Whether the reduction of vascular complications may impact survival remains to be demonstrated in appropriate prospective studies. Furthermore, we found two additional independent predictors of mortality in thrombosis-mediated transition, namely platelet count >1000 × 109/L (HR = 5.74, 95% CI = 1.79–18.40, p = 0.003), in line with a previous observation [13], and the incident arterial vs. venous thrombosis (HR = 4.43, 95% CI = 1.04–18.91, p = 0.044).
Limitations of this study concern its retrospective design and a possible bias related to the reporting accuracy of events, in terms of completeness and timing. In addition, since in these databases, the administration times of the cytoreductive drugs (hydroxyurea in absolute prevalence) were not well specified, we could not reliably evaluate the influence of the pharmacological cytoreduction on the post-diagnosis events. Furthermore, given that current results were obtained in the same ET database used for IPSET scores, a possible “self” confirmation bias could not be excluded. However, our aim was not to confirm the overall performance of the two scores, but to evaluate whether the transition from one state to another could have affected the overall survival or the cumulative incidence of thrombosis in a different way.
Strengths of the study are the relatively large number of patients for rare diseases such as ET and pre-PMF and the clinical and hematological diagnostic accuracy of the two entities and outcomes.
In conclusion, this multistate analysis provides novel information on the temporal probability of intermediate critical events occurring in ET and pre-PMF, and their impact on mortality. This knowledge might inform clinical practice and could also make more feasible the design of clinical trials.
Helna Pettersson, Jenni Adamsson, Peter Johansson, Staffan Nilsson, Lars Palmqvist, Bjorn Andreasson, Julia Asp
Introduction: Myeloproliferative neoplasm (MPN) is a heterogenous group of hematological malignancies including polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). JAK2V617F is the most frequent driver mutation in all three entities, but in PMF and ET mutations in CALR and MPL are also frequent. Mutations seen in additional genes are also often the same regardless of subtype of MPN. The aim of this study was to analyze a population based MPN cohort for genetic variants with prognostic value that can guide clinical decisions.
Methods: MPN patients from Western Sweden diagnosed between 2008-2013 (n=248) were screened for mutations in 54 genes associated with myeloid malignancy.
Results: Mutations in the genes SRSF2 and U2AF1 correlated significantly with impaired overall survival but did not correlate to increased risk for vascular events, neither before nor after diagnosis. Rather, mutations in these genes showed an association with disease transformation. Several recurrent gene variants with allele frequency close to 50% were confirmed to be germline. However, none of these variants was found to have an earlier onset of MPN.
Discussion: In conclusion, we identified gene mutations to be independent markers of impaired survival in MPN. This indicates the need for more individualized assessment and treatment of MPN patients and a wider gene mutation screening already at diagnosis. This could ensure the identification of patients with high-risk mutations early on. In addition, several genetic variants were also identified as germline in this study but gave no obvious clinical relevance. To avoid conclusions from non-informative genetic variants, a simultaneous analysis of normal cell DNA from patients at diagnosis should be considered.
Introduction
Polycythemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF) all belong to the Philadelphia chromosome negative myeloproliferative neoplasm (MPN) category. These three entities share the same characteristics of causing proliferation of bone marrow cells, resulting in an increase of blood cells of myeloid lineage in the bone marrow and in peripheral blood. Advanced stages of PMF, on the other hand, is characterized by increase of reticulin fibers leading to decreased blood cells (1–3). The complications of these three entities are also similar regarding vascular events, i.e., thrombosis and bleeding. Furthermore, all three entities can transform into acute leukemia and have an impact on survival, however with large differences in frequency. Despite the common driver mutations in JAK2, CALR and MPL (4), the clinical presentation, risk and frequencies of complications and survival differ wildly between individual patients. Prognostic tools are therefore desired in clinical practice for follow-up and treatment decisions already at diagnosis. Modern sequencing techniques have given the opportunity to simultaneously analyze several mutations in blood malignancies. It has become widely used in both research and clinical practice (5). We and other groups have published data on risk mutations using this approach, but several studies analyze data on separate MPN entities or driver mutation groups. Since both driver mutations and several of the additional mutations found are shared between the subtypes of MPN, and since the occurrence of some additional mutations are rather rare, we hypothesize that analysis of mutations in MPN as one group has a potential to extend the prognostic value of genetic markers. Furthermore, there is a growing number of hereditary gene variants that have been linked to predisposition for development of hematological disorders, including MPN (6–11). This also represents a challenge when analyzing large amount of sequencing data, especially if comparison with normal non-malignant cells is not available. We also need to get more information regarding gene variants of unknown significance, to avoid overestimation of their importance but also to identify variants that might influence disease development and prognosis. In this study, we analyzed a well-defined, population based MPN cohort, regardless of subtype, for genetic variants. The aim was to search for additional prognostic markers that can be used to guide clinical decisions, as well as to investigate the potential impact of germline variants detected in the sequence data.
Materials and methods
Patients
All patients diagnosed with PV, ET or PMF according to the 2008 WHO diagnostic criteria (3) in Western Sweden at the Sahlgrenska University Hospital or NU Hospital Group between 2008 and 2013 and reported to the Swedish national blood cancer registry were identified. Basically, all patients in our health care region with suspected MPN are referred to and treated at these two hospitals. Thus, this cohort cover patients in the geographic area without any selection. Of this cohort, 248 patients were included in the study based on informed consent, availability of DNA sample from the time of diagnosis, and review of diagnosis. Data from a subset of this patient cohort have been published previously (12, 13). Details are outlined in Supplementary Table 1. Clinical characteristics, clinical course, vitality status, vascular complications, disease transformation and co-existing cancers were collected from the medical records of all patients. Each of the patient’s hospital records were searched after emergency care consultation and hospital admission records that is related to bleeding or thrombotic complications. Follow-up was done from diagnosis until June 2021. The study was performed in accordance with the Declaration of Helsinki after ethical approval.
Screening for myeloid mutations
Genomic DNA from whole blood from the same sample that was analyzed at diagnosis for the presence of JAK2, CALR or MPL mutation was screened for gene variants in 54 genes or mutational hot spots associated with myeloid malignancies. The TruSight Myeloid Sequencing panel (Illumina FC-130-1010) which also was used for diagnostics in the clinical laboratory at the time of the study, was used, and sequencing was performed on a MiSeq instrument (Illumina) according to manufacturer’s instructions. Secondary analysis was performed with MiSeq Reporter, v.2.4.60.8, using Burrows-Wheeler Aligner mapper and somatic variant caller (Illumina). Data were filtered and mapped to the human genome reference hg19 using Variant studio v3.0 (Illumina), where global filtering was set to >3% and coverage had a minimum of 500 reads. Variants causing missense, frameshift, an altered stop/initiation codon, in-frame insertion/deletion or variants affecting splice site were regarded as mutations. Variants with quality >Q30 and allele frequencies of at least 5% were considered positive for mutation. Known sequencing artefacts and variants previously found in normal controls were excluded from further analysis according to filter strategies used in the clinical laboratory. BAM files from secondary analysis were used to analyze selected variants by Integrative Genomics Viewer (www.broadinstitute.org). Variants in areas with difficult reads were excluded. Previously analyzed data was reanalyzed according to updated bioinformatic settings to make the results comparable regardless of time for sequencing.
Confirmation of germline variants
Blood sample was taken from patients with variants in CDKN2A (rs3731249), ETV6 (rs145477191), NOTCH1 (rs61751489) or MPL (rs41269541), with a variant allele frequency close to 50%. Blood was enriched for CD3+ cells, using MACS® cell separation kit StraightFrom™ Whole Blood CD3 MicroBeads (Miltenyi Biotech) and Whole Blood Column Kit (Miltenyi Biotech), according to the manufacturer’s protocol. Genomic DNA from CD3+ enriched cells were extracted using QIAamp DNA Blood Mini Kit (Qiagen) and 10 ng of DNA were genotyped using TaqMan SNP genotyping assay (Applied biosciences, Life technologies) according to manufacturer’s protocol. The following assays were used: CDKN2A (assay ID: C_25611114_10), ETV6 (assay ID: C_162058060_10), NOTCH1 (assay ID: C_90123839_10) and MPL (assay ID: ANFV4EK). All samples were analyzed in triplicates using the QuantStudio 3 Real-Time PCR system (ThermoFisher Scientific). Genotypes were determined automatically based on dye component fluorescent emission data depicted in the X–Y scatter plot using Taqman genotyper software v.1.6.0. The gnomAD database v2.1.1 (https://gnomad.broadinstitute.org/) was used to compare frequencies in the MPN cohort with a normal Swedish population.
Statistical analysis
Fisher’s Exact Test was used to compare differences in frequencies between groups. To estimate overall survival (OS), defined as time from diagnosis to last follow up or death from any cause, the Kaplan Meier Log-rank test was used initially. For multivariable analysis, logistic regression and Cox Regression was used. P-values <0.05 were considered statistically significant. The statistical software used were Analyze-it v.6.15.4 (Microsoft Excel), GraphPad Prism v.9.4.0 and SPSS v29.0.0.0.
Results
A population-based cohort
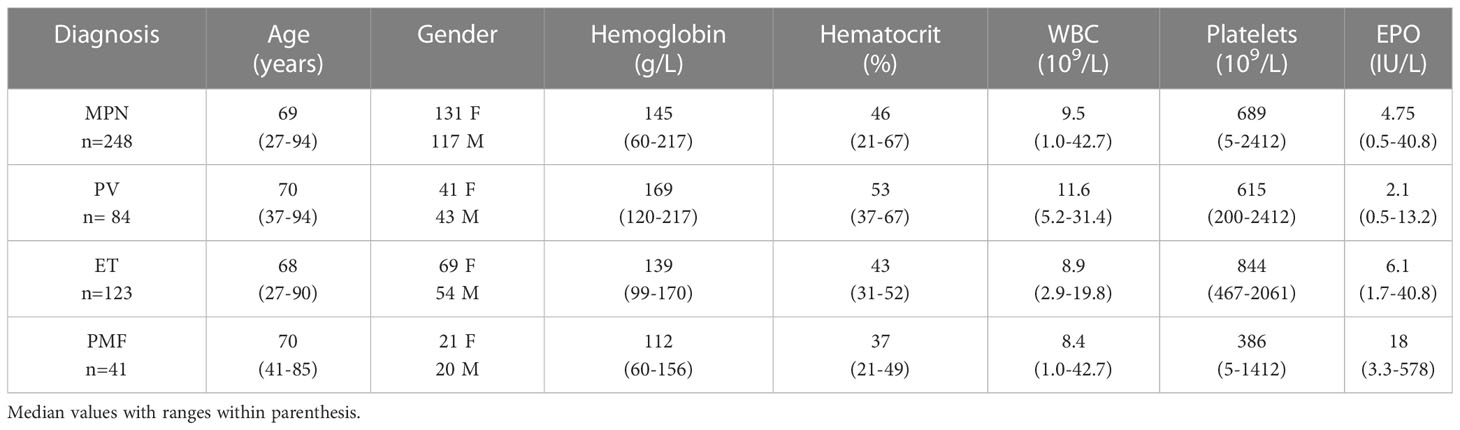
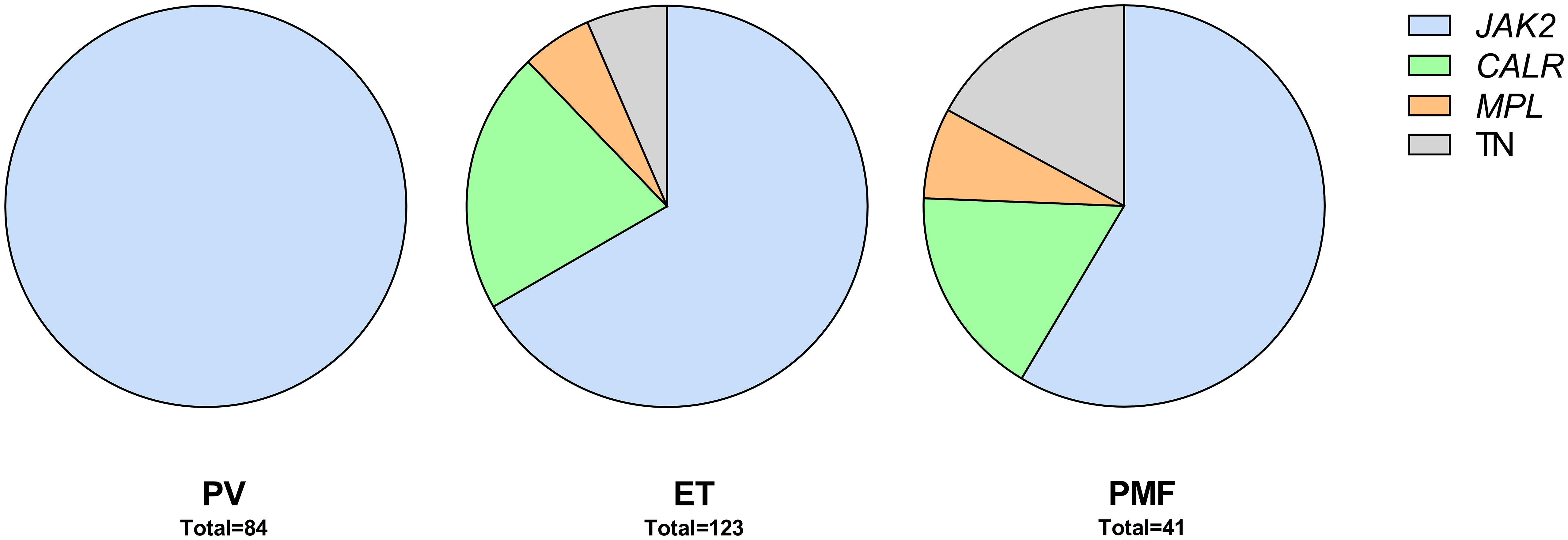
Between 2008 and 2013, 300 patients were diagnosed with MPN at Sahlgrenska University Hospital and NU Hospital Group in Western Sweden. Of these, 83% (n=248; PV n=84, ET n=123, PMF n=41) were included in the study. All included patients fulfilled the 2008 WHO diagnostic criteria. Age, gender, and blood counts from the time of diagnosis, for the whole MPN group and for the sub entities, are presented in Table 1. The distribution of driver mutations found at diagnosis was consistent with expected findings in the different subgroups of MPN (Figure 1). One patient with PMF was found to harbor both JAK2 V617F as well as mutation in the CALR gene. Patients not included in the study either declined to participate, had another diagnosis when their medical records were reviewed, or diagnostic material was missing. The median age of these patients was slightly lower (66 years vs. 69 years) but there was no other significant difference between these and the included patients.
Table 1
Table 1 Age, gender and laboratory findings at diagnosis in 248 patients with MPN.
Figure 1
Figure 1 Patients by diagnosis and driver mutations. TN, triple negative.
Mutations and survival
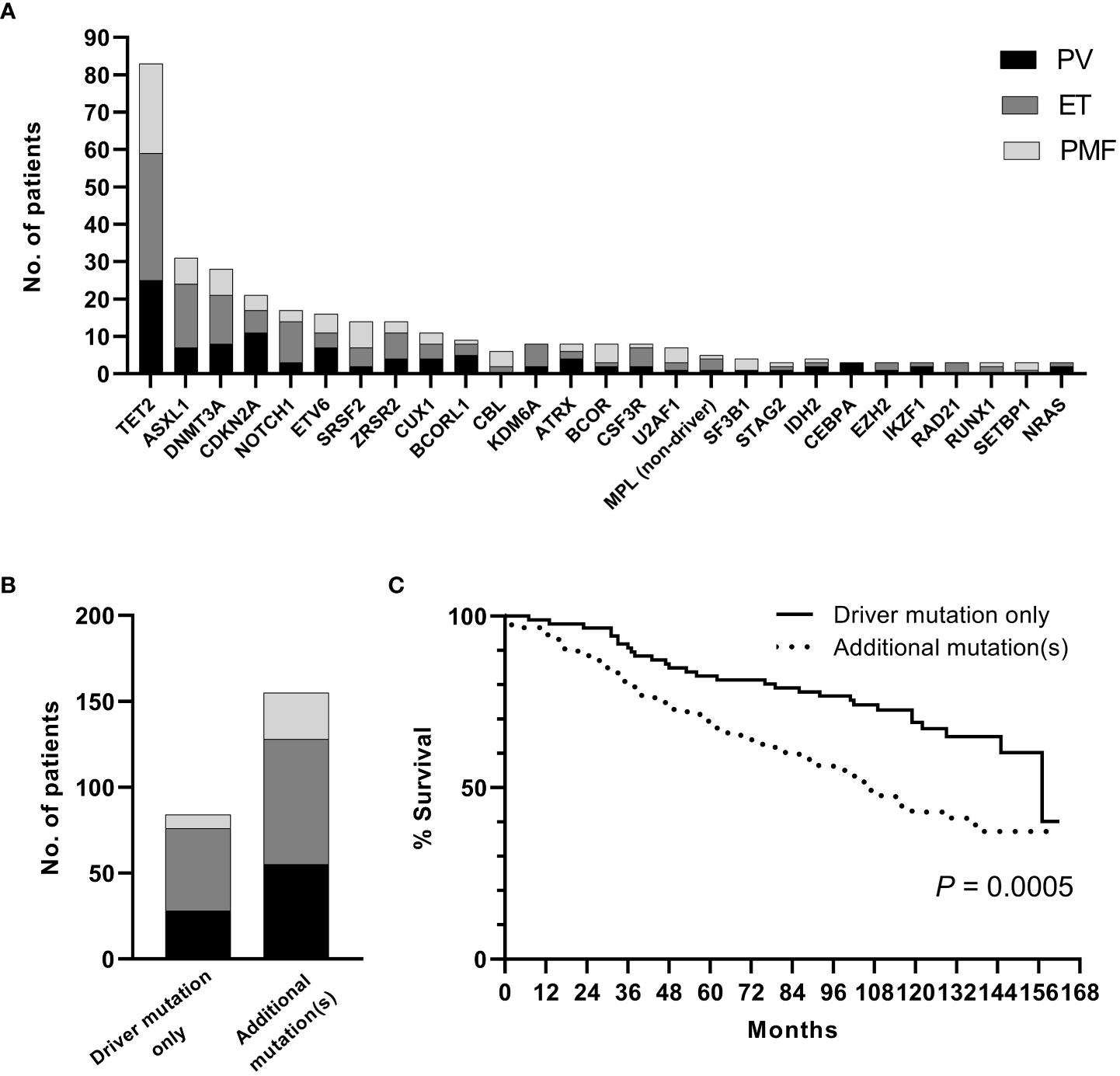
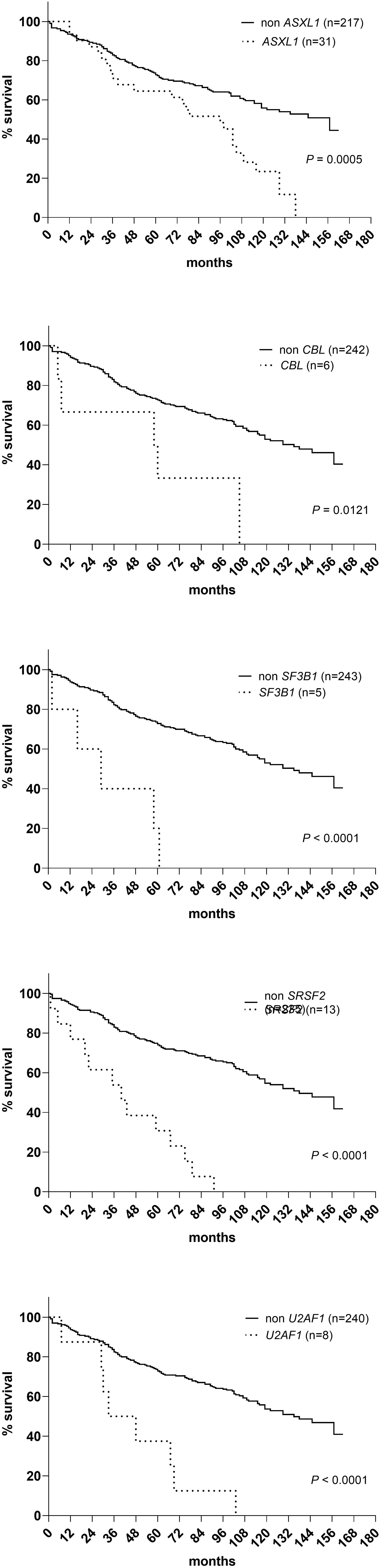
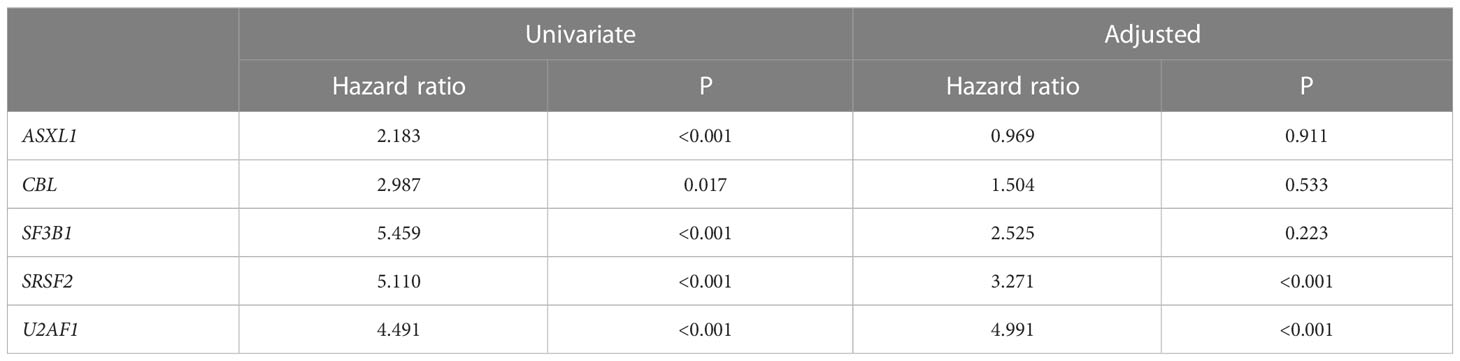
A sequencing panel including 54 genes associated with myeloid malignancies was used to screen for genetic variants that could be used as prognostic markers. Variants regarded as mutations other than the diagnostic driver mutations (JAK2, CALR or MPL) were found in 37 genes in at least one patient and in 27 genes in at least three patients (Figure 2A; Supplementary Table 1). During analysis of the gene data, several recurrent gene variants with allele frequency close to 50% were noted, which implied a hereditary variant. Therefore, analysis of the most common variants CDKN2A (NM_001195132.1:c.442G>A), NOTCH1 (NM_017617.3:c.6853G>A) and ETV6 (NM_001987.4:c.602T>C) as well as a variant close to a splice site in the MPL gene (NM_005373.2:c.1565 + 5C>T) were also analyzed in separated T-cells from new blood samples. This confirmed the variants to be germline. Therefore, these variants were excluded from analyses of prognostic impact. Sixty-three percent of the MPN cases had other mutations in addition to the diagnostic driver mutation (Figure 2B). Presence of at least one additional mutation was found to be associated with inferior survival (Figure 2C). To investigate if it was mutations in general or mutations in particular genes that had impact on survival, all genes with mutations detected in at least three patients were correlated to survival with the Kaplan Meier Log-rank test. For the whole MPN group, only mutations in five genes correlated significantly with inferior overall survival, ASXL1 (P=0.0005), SRSF2 (P<0.0001), U2AF1 (P<0.0001), CBL (P=0.01) and SF3B1 (P<0.0001) (Figure 3). These were further tested with multivariable analysis using Cox regression where also age and type of diagnosis were taken into consideration. When the five genes were grouped together, they still correlated to OS (P=0.002) with a hazard ratio 3.248, and it was not dependent on type of diagnosis (interaction 0.592). Also, age at diagnosis correlated to OS (P<0.001) as expected. However, it should be noted that all cases with CBL mutation also harbored mutation in another of the four genes (Supplementary Table 1). Therefore, the genes were also tested separately. When these were adjusted for both age and type of diagnosis, only mutations in SRSF2 and U2AF1 correlated significantly to OS (Table 2).
Figure 2
Figure 2(A). Frequency of additional mutations. (B). Distribution of patients with driver mutation only or addition mutation(s) when confirmed germline variants have been excluded. (C). OS in patients with driver mutation only or with addition mutation(s) (confirmed germline variants excluded).
Figure 3
Figure 3 OS in patients with mutations in ASXL1, CBL, SF3B1, SRSF2 and U2AF1 respectively according to Kaplan Meier Log-rank test.
Table 2
Table 2 Mutation impact on OS in univariate or adjusted (including age and type of diagnosis) analysis.
Mutations and vascular events
Vascular events in MPN are potentially life-threatening. The vascular complications are either thrombosis or bleeding where the co-existence of MPN is a contributing factor. The most common incidences are those that are discovered at the time of diagnosis and the most common thrombotic events were myocardial infarction (n=27), cerebrovascular infarction (n=24), pulmonary embolism (n=19), transient ischemic attack (n=10) and deep vein thrombosis (n=8). The most frequent hemorrhagic complications were gastro-intestinal (n=11) and cerebral bleedings (n=6). Fisher’s Exact Test and logistic regression were used to analyze if SRSF2 or U2AF1 which correlated with shorter OS also correlated with occurrence of vascular events before or after diagnosis or in total. However, no such correlation was seen, neither when only the mutated genes were tested, nor when they were combined with age and type of diagnosis.
Mutations and disease transformation
All MPNs have a risk of transformation into secondary acute myeloid leukemia (AML). In our cohort, 17 cases had transformed to AML. Both mutated genes with correlation to OS were tested with logistic regression. This showed that mutations in SRSF2 correlated with AML transformation (P=0.002), but this was not the case for U2AF1 (P=0.236). The analysis was extended to find genes correlated to fibrotic transformation and co-existence with other myeloid hematological malignancies. There were 18 patients that had secondary myelofibrosis transformation from PV and ET. Other myeloid hematological malignancies that co-existed with MPNs included two chronic myelomonocytic leukemia and one myelodysplastic syndrome. Logistic regression showed that mutations in both SRSF2 and U2AF1 correlated with co-existing myeloid hematological malignancies (SRSF2 P=0.05 and U2AF1 P=0.014).
Gene germline variants
Identified germ line variants indicated a possible hereditary predisposition of MPN. Comparison of the frequency in our MPN cohort to a normal Swedish population cohort in the gnomAD variant database was performed. Only the variant found in ETV6 was more frequent in the MPN group (0.0282 vs. 0.00975 in allele frequency). This difference was statistically significant (Fischer’s exact test, p=0.0006). However, there was no correlation between any of the variants and occurrence of early onset MPN. We further used logistic regression to test if any of the variants correlated with occurrence of other cancers (both solid tumors and hematological malignancies outside the MPN group). These cancers occurred about the time and after diagnosis of MPN and were noted upon reviewing the patient´s hospital records. In total, 19 patients with non-hematologic cancers were found. The most common types were colon cancer (n=6) and pancreatic cancer (n=4). However, no significant correlation was seen.
Discussion
The serious risks all MPNs impose, although at various frequencies, are vascular complications, transformation to more severe hematologic malignancies and ultimately negative impact on OS. It is thus a priority to identify high risk patients in clinical practice. Age at diagnosis as well as occurrence of vascular complications have been reported as risk factors (14, 15). Access to an abundance of genetic data allows genetic profiling to further broaden prognostic information. Mutational status has progressively taken a big role in clinical practice. Occurrence of mutations have also been used to create scoring systems for MPN (16, 17). The initial focus on gene mutations in MPN was on the driver mutations’ importance on disease development. These mutations are found in the genes JAK2, CALR and MPL which all are involved in JAK-STAT signaling (18). Notably, the same JAK2 mutation is found in both PV, ET and PMF, and mutations in CALR and MPL are seen in both ET and PMF. Thus, the mutation itself does not seem to determine the MPN phenotype, instead, allele burden has been reported as one factor behind the phenotypic differences (19). The order of acquisition of the driver mutation in relation to additional mutations may also have influence (20, 21). If the JAK2 mutation precedes mutation in DNMT3A or TET2, the phenotypic picture would likely be PV. If mutations instead occur in reverse order, the MPN phenotype would likely be ET (22). Host factors also contribute to the development of disease (22, 23). Several predisposing gene variants have been identified that may influence not only the risk of developing disease but also the course of the disease (17, 24–26). It is therefore reasonable to investigate the whole MPN cohort as a group independent of diagnosis when analyzing it from a genetic point of view.
Aside from the driver mutations, several additional mutations have also been reported in MPN. These are subclassified to gene families: epigenetic regulators (ASXL1, EZH2, TET2, IDH1/2, DNMT3A), spliceosome (SRSF2, SF3B1, U2AF1, ZRSR2), transcriptional regulators (TP53, RUNX1, IKZF1), general cell signaling genes (KRAS, PTPN11) as well as specific negative regulators of JAK/STAT signaling (CBL) (27). Occurrence of additional mutations correlated significantly with inferior OS (Figure 2C). Genetic profiling has raised the question if the number of mutations in a particular case is more important for the occurrence of complications or OS rather than in what genes or type of gene the mutations are present. Our results suggest it is not the number of mutations but rather the presence of certain gene mutations that are more informative for prognostic guidance. Mutations in SRSF2 and U2AF1 correlated significantly with worse OS in our patient cohort. Although they were more frequent in PMF, which is well known to have an impaired survival compared to patients with PV and ET, mutations in these two genes correlated to worse OS regardless of MPN subtype.
Previous studies, including a subpopulation of our analyzed MPN cohort, have shown that triple negative MPN without JAK2, CALR or MPL mutation have worse prognosis (12, 28). It has also been shown that the presence of several other mutations in addition to a driver mutation correlate with survival (2, 5, 24, 26, 28–32). In this study we initially identified mutations in five non-driver genes (ASXL1, SRSF2, U2AF1, SF3B1 and CBL) to be significantly correlated to impaired OS. Mutations in both ASXL1 and SRSF2 have previously been classified as high-risk mutations in both PMF and PV (26, 33). Moreover, mutations in U2AF1 and SF3B1 have been identified as genetic risk factors in ET (17). When age at diagnosis as well as type of diagnosis was taken into consideration only mutations in SRSF2 and U2AF1 remained associated with shorter OS. Mutations in CBL were only found in those patients who harbored mutations in one or more of the four other genes, suggesting that mutated CBL might just be a passenger rather than a disease driver. Mutations in ASXL1 is commonly seen in clonal hematopoiesis, which increases with age (34). This could be an explanation why the presence of ASXL1 mutation no longer significantly correlated with OS when age was taken into consideration.
Since vascular complications are associated with impaired survival we wanted to investigate if the detected mutations correlated also to vascular events in our MPN cohort. However, neither mutations in SRSF2 nor U2AF1 correlated to vascular events before or after diagnosis or in total. Another complication with MPN is transformation to myelofibrosis for PV and ET or to secondary AML for all three MPN. In the present cohort, a significant correlation between mutations in SRSF2 and transformation to AML was found. Furthermore, mutations in both SRSF2 and U2AF1 correlated with transformation from PV and ET to myelofibrosis and development of other hematological malignancies. This is in line with previous findings were mutations in SRSF2 and U2AF1 have been reported to serve as prognostic markers for rapid blastic progression in newly diagnosed MPN (35). Moreover, mutations in SRSF2, U2AF1 and SF3B1 detected at presentation of disease have been associated with rapid fibrotic progression in PMF. This was not demonstrated for mutations in ASXL1, DNMT3A or TET2 (36).
Aside from the acquired mutations in our MPN cohort, several specific variants were identified which turned out to be germline. A five- to sevenfold higher risk of MPN among first-degree relatives to MPN patients have previously been reported in Sweden, which suggest a genetic predisposition (37). Also in other myeloid malignancies, the question for germline variants involved in disease have come into focus (38–40). Four variants in our study were more closely investigated, their allele frequency was close to 50%, which could imply a hereditary variant. These genes were CDKN2A (NM_001195132.1:c.442G>A), NOTCH1 (NM_017617.3:c.6853G>A) and ETV6 (NM_001987.4:c.602T>C) as well as a variant close to a splice site in the MPL gene (NM_005373.2:c.1565 + 5C>T). The most frequent occurring CDKN2A mutation leading to a p.A148T substitution has been reported as an inherited coding variant associated with leukemic transformation of hematopoietic progenitor cells (41). Comparison of the frequency to a normal Swedish population cohort, however, only revealed the ETV6 variant to be more common in the MPN patient cohort. This variant did not correlate to earlier onset of disease, which could be expected for an inherited predisposition. On the other hand, in the Landgren study the mean age at diagnosis did not differ between affected relatives and controls (37). The ethical approval of the current study did not include testing of relatives, but it would of course be of interest to see if any of these variants are associated with an increased incidence of hematological or non-hematological malignancies within these families. Nevertheless, it is important to correctly identify germline gene variants to avoid drawing conclusions from non-informative genetic variants but also to provide genetic counseling when called for. In this study we used CD3+ selection of T-cells from collected blood samples to get constitutive DNA, which turned out to be easiest for both referring doctors and gave acceptable DNA yield for the laboratory but might of course misdiagnose somatic variants that are also present in lymphoid cells. Another alternative is a skin biopsy but this may be considered too much of an intervention for some patients.
In conclusion, our study on a population based MPN cohort strengthens previous reports about prognostic value of genetic data in MPN. Thus, a wider gene profiling at diagnosis is of value. In addition, several genetic variants were also identified as germline in this study but gave no obvious clinical relevance. To avoid conclusions from non-informative genetic variants, simultaneous analysis of normal cell DNA from patients at diagnosis should be considered.
Myelofibrosis is a rare type of bone marrow cancer. In this condition, extensive scarring (fibrosis) occurs in the bone marrow, which keeps the bone marrow from producing the right number of blood cells.1
Healthy functioning bone marrow produces infection-fighting white blood cells (WBCs), oxygen-carrying red blood cells (RBCs), and blood-clotting platelets in the correct numbers the body needs to function normally.
When myelofibrosis occurs, some people may not have any symptoms, while others have severe symptoms that require immediate treatment. This article will explain the symptoms of myelofibrosis, how it is diagnosed, and how it is treated.
How Myelofibrosis Affects the Body
Bone marrow is present inside the middle of the bones. It is normally a soft, spongy texture. It produces WBCs, RBCs, and platelets.
In myelofibrosis, one of the cells of the bone marrow begins to grow abnormally, multiply, and continue to produce more abnormal cells. Eventually, abnormal cells are present in high enough numbers to crowd out the healthy cells.
These abnormal cells cause fibrosis, which prevents the bone marrow from producing the correct number of blood cells the body needs to function normally.2 Over time, there is an increased risk of developing acute myeloid leukemia (AML), another form of blood cancer.1
Myelofibrosis Types
The initial cause of abnormal bone marrow development determines the type of myelofibrosis. The two types are primary and secondary.
Primary Myelofibrosis
With primary myelofibrosis, the change in the bone marrow cells happens spontaneously. It doesn’t occur due to a previous bone marrow condition.1
Secondary Myelofibrosis
With secondary myelofibrosis, the fibrosis occurs due to another bone marrow disorder, specifically polycythemia vera or essential thrombocythemia.3
Polycythemia vera is a blood disorder in which the bone marrow produces too many blood cells, most often red blood cells, but can also include white blood cells and platelets.2 Essential thrombocythemia is a disorder in which the bone marrow makes too many platelets.4
Myelofibrosis Symptoms
As too few blood cells are made, symptoms will start to develop. The rate at which symptoms develop and how severe they become can vary from person to person, and may take years to be experienced. Symptoms associated with myelofibrosis include:2
Feeling tired
Shortness of breath
Pale skin
Headaches
Fever
Night sweats
Enlarged spleen
Enlarged liver
Frequent infections
Easy bleeding or bruising
Abdominal pain
Joint pain
Bone pain
Tumors may develop in the lungs, skin, liver, or gastrointestinal tract and cause further symptoms.2
Causes of Myelofibrosis
For those living with primary myelofibrosis, the exact cause of the disease may never be known.
However, in about half of the cases of primary myelofibrosis, a mutation in the JAK2gene is found.5 The JAK2 mutation is also frequently found in those with polycythemia vera and essential thrombocythemia.
This mutation isn’t inherited. Instead, it develops spontaneously in a bone marrow cell. It produces a protein that causes the bone marrow to overproduce platelet precursor cells called megakaryocytes. These cells stimulate other cells to produce too much collagen, a protein that then builds up and produces scarring in the bone marrow.
Other gene mutations that may play a role in developing myelofibrosis include the CALR and MPL genes.1
Risk factors that may play a role in developing primary myelofibrosis include:2
Increasing age
History of exposure to chemicals including benzene, fluoride, or phosphorus
Risk factors for developing secondary myelofibrosis include:6
Having another cancer that has spread into the bone marrow
Having polycythemia vera or essential thrombocythemia
Diagnosis of Myelofibrosis
The diagnosis of myelofibrosis often starts when someone presents to their healthcare provider for evaluation of a symptom that they are experiencing. During this evaluation, a healthcare provider may start with a detailed history and physical examination. Blood work may be taken which can start the process of finding a diagnosis.1
A complete blood count (CBC) and peripheral blood smear measure the number of WBCs, RBCs, and platelets, as well as their shape and size. Abnormal findings in the CBC may lead to further testing, which may include a bone marrow biopsy.1
During a bone marrow biopsy, a small sample of bone marrow is taken, often from the hip. This allows the pathologist (physician specializing in analyzing body fluids and tissues in a lab setting) the ability to evaluate for any changes or abnormalities in the bone marrow. This test can result in a diagnosis of myelofibrosis.
In addition, a physical exam or imaging study such as a computed tomography (CT) scan may reveal an enlarged spleen.
Other blood testing may include:1
Complete metabolic panel (CMP) to evaluate kidney and liver function
Coagulation studies
Iron levels
Lactate dehydrogenase (LDH) to assess inflammation and tissue damage
Testing for the JAK2, CALR, and MPL gene mutations
Once a diagnosis of myelofibrosis is made, it is further classified into different risk categories, which helps determine how likely the disease is to turn into AML and can help determine treatment options.
This score is determined by the person’s age, symptoms, hemoglobin level, platelet count, leukocyte (a white blood cell) and leukoblast (a developing white blood cell) count, and certain genetic changes. The higher the score, the more high-risk their myelofibrosis is.
Myelofibrosis Treatment
Some people diagnosed with myelofibrosis, especially those without many symptoms or who have low-risk disease, may not receive any treatment until they become symptomatic. Called a watchful waiting approach, this policy of taking no immediate action regarding treatment includes routine blood tests and visits with their healthcare provider to determine when treatment will be needed.2
If someone is experiencing symptomatic anemia (low RBCs) because of myelofibrosis, they may receive periodic RBC transfusions. There are additional medications that may be given to help the bone marrow make red blood cells.
This may not completely resolve anemia but can keep the red blood cells up at a tolerable level. If someone also has iron deficiency anemia, iron supplements may help improve red blood cell levels.2
To reduce high levels of WBCs and platelets, medications to suppress the bone marrow may be given. An example of one of these medications is hydroxyurea.
An enlarged spleen may need to be treated if it is contributing to symptoms, especially pain or severely low platelets. This can be done through radiation to the spleen or by surgical removal of the spleen.
A medication called Jakafi (ruxolitinib) can be prescribed to those with either primary or secondary myelofibrosis who fall in the moderate- or high-risk category. This medication interferes with the JAK2 pathway that the cells use to grow. Another medication, Inrebic (fedratinib) can also be used to treat intermediate or high-risk primary or secondary myelofibrosis.2
Can Myelofibrosis Be Cured?
The majority of cases of myelofibrosis are treated with the goal of decreasing symptoms of the disease. An attempt can be made to cure the disease through a stem cell transplant.
This approach requires large doses of chemotherapy to kill all of the cancer cells. Stem cells are collected before the procedure to be infused back in after chemotherapy has worked. These stem cells can then begin to resume making normal WBCs, RBCs, and platelets. This procedure is not recommended for everyone with myelofibrosis, as it can lead to severe complications.7
Complications Associated With Myelofibrosis
Complications associated with myelofibrosis are related to the severe decrease in the number of normal WBCs, RBCs, and platelets. As the disease progresses and the blood counts continue to decrease, complications may arise.
With the decrease of white blood cells comes a higher risk of developing infection. Infections can occur anywhere in the body, though most often in the lungs. The infection can be due to bacteria, viruses, or fungi. With infection may come fever, increased weakness, and cough.8
Low red blood cells can result in severe anemia, which prevents enough oxygen-rich blood from getting to the tissues in the body. With the decreased amount of available oxygen comes complications such as heart failure, in which the heart has to work too hard to try to keep up with the increased demand for oxygen.8
Not having enough normal platelets can lead to severe bleeding or hemorrhaging. The bleeding can occur following an injury or can occur spontaneously. The bleeding can become life-threatening if severe and not stopped quickly.8
Blood clotting, the opposite of bleeding, could also occur. If blood clots inappropriately, it can lead to clots moving around the body and getting stuck in areas they are not supposed to be. This can lead to damage to the brain, heart, lungs, and extremities.
Transformation to acute myeloid leukemia occurs in 5% to 10% of those diagnosed with myelofibrosis. This is most common in primary myelofibrosis and is a significant complication since the prognosis is poor with transformation into AML.9
Myelofibrosis Prognosis
The prognosis of myelofibrosis can vary from person to person. It depends upon the type and risk category of their disease. Prognosis can differ slightly based on which scale is used at the time of diagnosis. The table below references the prognosis scale MIPSS70, which is used for those 70 years old or younger and is based on risk group severity.9
Risk Group
10-Year Survival
Very high
Less than 5%
High
13%
Intermediate
37%
Low
56%
Very Low
92%
When to Contact a Healthcare Provider
See a healthcare provider if you are having symptoms associated with myelofibrosis. Many of these are also associated with other conditions. A workup and diagnosis can ensure you are getting the appropriate treatment.
If you have been diagnosed with myelofibrosis, contact your healthcare provider anytime you’re having concerns about the symptoms you’re experiencing, especially if they continue for some time without getting better.
Your provider may want to run additional tests or start treatment if your symptoms continue. If severe symptoms develop, notify your healthcare provider immediately or seek emergency care.
Summary
Myelofibrosis is a type of blood cancer in which abnormal cells cause the bone marrow to become extensively scarred (fibrosis). The fibrosis doesn’t allow the bone marrow to make blood cells properly, which leads to low blood counts and other complications.
Once formally diagnosed by a bone marrow biopsy, the results will be used by your healthcare provider to develop a treatment plan. Treatments are individualized, ranging from watchful waiting to stem cell transplant.